This tutorial focuses on aggregating and combining various climate and phenology
data sources for modeling purposes using the phenor R package. This tutorial
explains the various data sources and in particular phenocam data, the structure
of the formatted data and the final modelling procedures using various phenology
models.
R Skill Level: Introduction - you've got the basics of R down and
understand the general structure of tabular data and lists.
Learning Objectives
After completing this tutorial, you will be able:
to download PhenoCam time series data
to process time series data into transition date products (phenological events)
to download colocated climate
to format these data in a standardized scheme
to use formatted data to calibrate phenology models
to make phenology predictions using forecast climate data
Things You’ll Need To Complete This Tutorial
You will need the most current version of R and RStudio loaded on your computer
to complete this tutorial. Optionally, a login to the
Pan European Phenology Project (PEP725)
website can be used for data retrieval.
Install R Packages
These R packages will be used in the tutorial below. Please make sure they are
installed prior to starting the tutorial.
devtoolsinstall.packages("devtools")
phenor:install_github("khufkens/phenor")
phenocamr:install.packages("phenocamr")
maps:install.packages("maps")
This tutorial has three parts:
Introductions to the relevant R packages
Aggregating & format the data
Model phenology
Due to the the large size of the data involved, we will learn how to obtain research
quality data in the aggregating data steps but we will use pre-subsetted data sets
for the modeling. The pre-subsetted sets can be downloaded at the end of each
section or directly accessed during the modeling section.
The R packages
phenor
The phenor R package is a phenology modeling framework in R. The framework
leverages measurements of vegetation phenology from four common phenology
observation datasets combined with (global) retrospective and projected climate
data. Currently, the package focuses on North America and Europe and relies
heavily on
Daymet
and
E-OBS climate data
for underlying climate driver data in model optimization. The package supports
global gridded CMIP5 forecasts for RCP4.5 and RCP8.5 climate change scenarios
using the
NASA Earth Exchange global downscaled daily projections.
Phenological model calibration and validation data are derived from four main sources:
the transition dates derived from PhenoCam time series and included in this package.
We will also use the the phenocamr package in the processing of data provided
through the PhenoCam API and past data releases. Although the uses of standard product
releases is encouraged in some instances you might want more control over the
data processing and the transition date products generated. phenocamr provides
this flexibility.
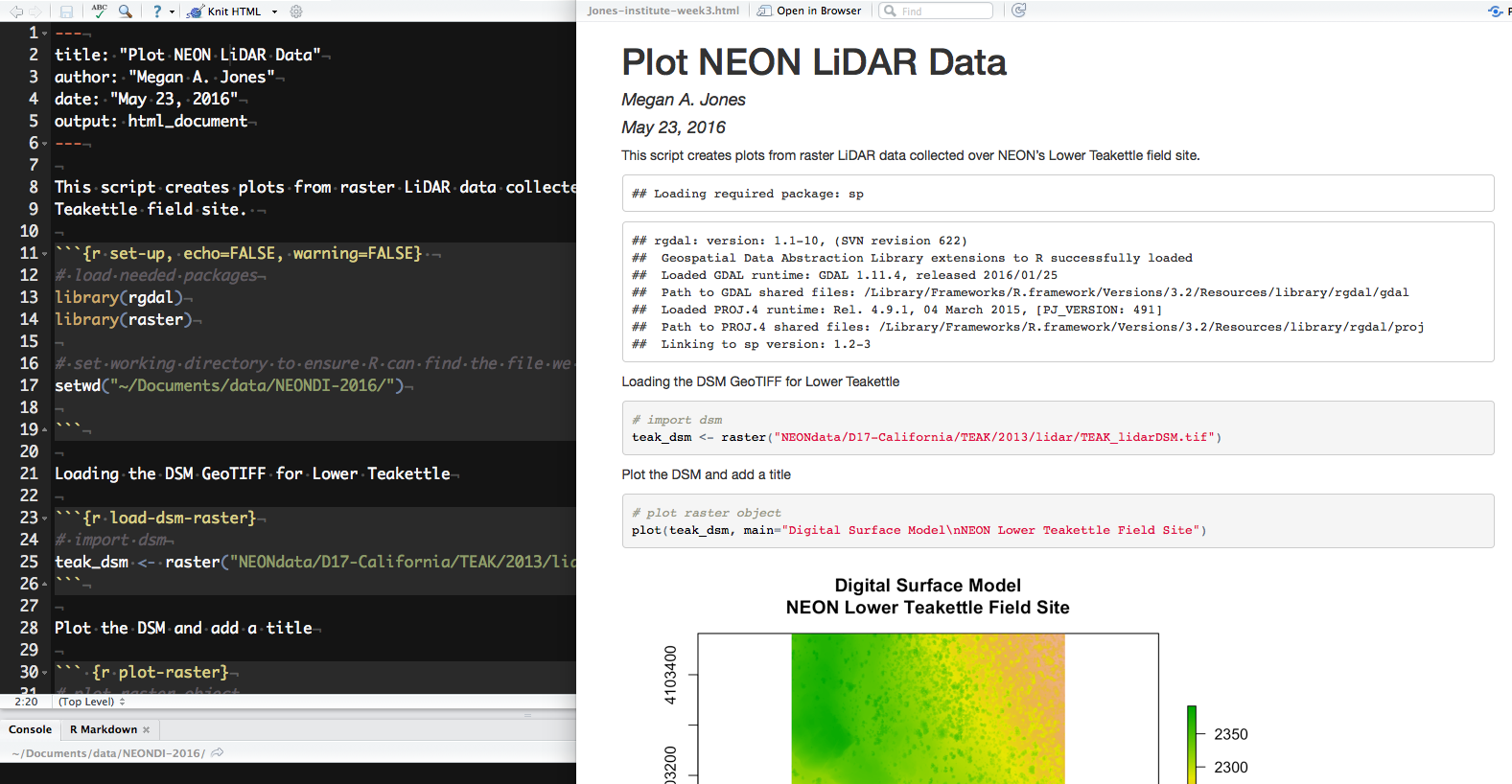
Get PhenoCam Data
In this tutorial, you are going to download PhenoCam time series, extract
transition dates and combine the derived spring phenology data, Daymet data, to
calibrate a spring phenology model. Finally, you make projections for the end
of the century under an RCP8.5 CMIP5 model scenario.
The PhenoCam Network includes data from around the globe
(map.)
However, there are other data sources that may be of interest including the Pan
European Phenology Project (PEP725). For more on accessing data from the PEP725,
please see the final section of this tutorial.
# download the three day time series for deciduous broadleaf data at the
# Bartlett site and will estimate the phenophases (spring + autumn).
phenocamr::download_phenocam(
frequency = 3,
veg_type = "DB",
roi_id = 1000,
site = "bartlettir",
phenophase = TRUE,
out_dir = "."
)
## Downloading: bartlettir_DB_1000_3day.csv
## -- Flagging outliers!
## -- Smoothing time series!
## -- Estimating transition dates!
Using the code (out_dir = ".") causes the downloaded data, both the 3-day time
series and the calculated transition dates, to be stored in your current working
directory. You can change that is you want to save it elsewhere. You will get feedback on the processing steps completed.
We can now load this data; both the time series and the transition files.
# load the time series data
df <- read.table("bartlettir_DB_1000_3day.csv", header = TRUE, sep = ",")
# read in the transition date file
td <- read.table("bartlettir_DB_1000_3day_transition_dates.csv",
header = TRUE,
sep = ",")
Threshold values
Now let's plot the data to see what we are working with. But first, let's
subset the transition date (td) for each year when 25% of the greenness amplitude (of the 90^th^) percentile is reached (threshold_25).
# select the rising (spring dates) for 25% threshold of Gcc 90
td <- td[td$direction == "rising" & td$gcc_value == "gcc_90",]
# create a simple line graph of the smooth Green Chromatic Coordinate (Gcc)
# and add points for transition dates
plot(as.Date(df$date), df$smooth_gcc_90, type = "l", xlab = "Date",
ylab = "Gcc (90th percentile)")
points(x = as.Date(td$transition_25, origin = "1970-01-01"),
y = td$threshold_25,
pch = 19,
col = "red")
Now we can se the transition date for each year of interest and the annual
patterns of the Gcc.
However, if you want more control over the parameters used during processing,
you can run through the three default processing steps as implemented in
download_phenocam() and set parameters manually.
Of particular interest is the option to specify your own threshold used in
determining transition dates. In the example below, we will set the upper
threshold value to 80% of the amplitude (or 0.8). We will visualize the data as
above, showing the newly found transition dates along the Gcc curve.
# the first step in phenocam processing is flagging of the outliers
# on the file you visualized in the previous step
detect_outliers("bartlettir_DB_1000_3day.csv",
out_dir = ".")
# the second step involves smoothing the data using an optimization approach
# we force the procedure as it will be skipped if smoothed data is already
# available
smooth_ts("bartlettir_DB_1000_3day.csv",
out_dir = ".",
force = TRUE)
# the third and final step is the generation of phenological transition dates
td <- phenophases("bartlettir_DB_1000_3day.csv",
internal = TRUE,
upper_thresh = 0.8)
Now we have manually set the parameters that were default for our first plot.
Note, that here is also a lower and a middle threshold parameter, the order matters so
always use the relevant parameter (for parameters, check transition_dates())
Now we can again plot the annual pattern with the transition dates.
# split out the rising (spring) component for Gcc 90th
td <- td$rising[td$rising$gcc_value == "gcc_90",]
# we can now visualize the upper threshold
plot(as.Date(df$date), df$smooth_gcc_90, type = "l",
xlab = "Date",
ylab = "Gcc (90th percentile)")
points(x = as.Date(td$transition_80, origin = "1970-01-01"),
y = td$threshold_80,
pch = 19,
col = "red")
With the above examples you can get a feeling of how to manually re-process
PhenoCam time series.
Phenocam Subsetted Data Set
To allow our models to run in a timely manner, we will use the subsetted data
that is included with the phenor packages for the modeling portion of this
tutorial. All deciduous broadleaf forest data in the PhenoCam V1.0 have been processed
using the above settings. This data set is called phenocam_DB.
In order to calibrate phenology models, additional climate data is required.
Some of this data is dynamically queried during the formatting of the data.
Alternatively, we can get climate data from another source, like the
Coupled Model Intercomparison Project (CMIP5).
The forecast CMIP5 data is gridded data which is too large to process dynamically.
In order to use the CMIP5 data to make phenology projections the data needs to
be downloaded one year at a time, and subset where possible to reduce file sizes.
Below you find the instructions to download the 2090 CMIP5 data for the RCP8.5
scenario of the MIROC5 model. The data will be stored in the R temporary
directory for later use. Please note that this is a large file (> 4 GB).
# download source cmip5 data into your temporary directory
# please note this is a large download: >4GB!
phenor::download_cmip5(
year = 2090,
path = tempdir(),
model = "MIROC5",
scenario = "rcp85"
)
phenor::download_cmip5(
year = 2010,
path = tempdir(),
model = "MIROC5",
scenario = "rcp85"
)
Format Phenology & Climate Data
If both phenology and climate data are available you can aggregate and format
the data for modeling purposes. All functions in the phenor package with a
format_ prefix serve this purpose, although some might lack phenology
validation data.
You can format phenocam data using the format_phenocam() function, which
requires you to provide the correct path to phenocam transition date files, like
those we downloaded above). This function will match the transition dates from
PhenoCam data with the appropriate Daymet data (dynamically).
In the next code chunk, we will format the phenocam transition date data
(in your working directory, ".") correctly. Then we will specify the direction of the curve to be considered and setting the Gcc percentile, threshold and temporal offset.
# Format the phenocam transition date data
# Specify the direction of the curve
# Specify the gcc percentile, threshold and the temporal offset
phenocam_data <- phenor::format_phenocam(
path = ".",
direction = "rising",
gcc_value = "gcc_90",
threshold = 50,
offset = 264,
internal = TRUE
)
## Processing 1 sites
##
# When internal = TRUE, the data will be returned to the R
# workspace, otherwise the data will be saved to disk.
# view data structure
str(phenocam_data)
## List of 1
## $ bartlettir:List of 13
## ..$ site : chr "bartlettir"
## ..$ location : num [1:2] 44.1 -71.3
## ..$ doy : int [1:365] -102 -101 -100 -99 -98 -97 -96 -95 -94 -93 ...
## ..$ ltm : num [1:365] 13.5 14.1 13.6 13 11.9 ...
## ..$ transition_dates: num [1:9] 133 129 122 133 130 128 136 130 138
## ..$ year : num [1:9] 2008 2009 2010 2011 2012 ...
## ..$ Ti : num [1:365, 1:9] 16 17.2 16.8 15.5 16.2 ...
## ..$ Tmini : num [1:365, 1:9] 7 10 10.5 7.5 6.5 11 16 14.5 7.5 3 ...
## ..$ Tmaxi : num [1:365, 1:9] 25 24.5 23 23.5 26 29 28.5 24 20 18 ...
## ..$ Li : num [1:365, 1:9] 11.9 11.9 11.8 11.8 11.7 ...
## ..$ Pi : num [1:365, 1:9] 0 0 0 0 0 0 5 6 0 0 ...
## ..$ VPDi : num [1:365, 1:9] 1000 1240 1280 1040 960 1320 1800 1640 1040 760 ...
## ..$ georeferencing : NULL
## - attr(*, "class")= chr "phenor_time_series_data"
As you can see, this formats a nested list of data. This nested list is consistent
across all format_ functions.
Finally, when making projections for the coming century you can use the
format_cmip5() function. This function does not rely on phenology data but
creates a consistent data structure so models can easily use this data.
In addition, there is the option to constrain the data, which is global,
spatially with an extent parameter. The extent is a vector with coordinates
defining the region of interest defined as xmin, xmax, ymin, ymax in latitude and
longitude.
This code has a large download size, we do not show the output of this code.
# format the cmip5 data
cmip5_2090 <- phenor::format_cmip5(
path = tempdir(),
year = 2090,
offset = 264,
model = "MIROC5",
scenario = "rcp85",
extent = c(-95, -65, 24, 50),
internal = FALSE
)
cmip5_2010 <- phenor::format_cmip5(
path = tempdir(),
year = 2010,
offset = 264,
model = "MIROC5",
scenario = "rcp85",
extent = c(-95, -65, 24, 50),
internal = FALSE
)
Climate Training Dataset
Given the large size of the climate projection data above, we will use subsetted
and formatted training dataset. In that section of the tutorial, we will directly
read the data into R.
Alternatively, you can download it here
as a zip file (128 MB)
or obtain the data by cloning the GitHub repository,
Now that we have the needed phenology and climate projection data, we can create our model.
Phenology Model Parameterization
Gathering all this data serves as input to a model calibration routine. This
routine tweaks parameters in the model specification in order to best fit the
response to the available phenology data using the colocated climate driver data.
The default optimization method uses Simulated Annealing to find optimal
parameter sets. Ideally the routine is run for >10K iterations (longer for
complex models). When the procedure ends, by default, a plot of the modeled ~ measured data
is provided in addition to model fit statistics. This gives you quick feedback
on model accuracy.
For the phenology data, we'll used the example data that comes with phenor. This
will allow our models to run faster than if we used all the data we downloaded
in the second part of this tutorial. phencam_DB includes a subset of the
deciduous broadleaf forest data in the PhenoCam V1.0. This has all been
processed using the settings we used above.
# load example data
data("phenocam_DB")
# Calibrate a simple Thermal Time (TT) model using simulated annealing
# for both the phenocam and PEP725 data. This routine might take some
# time to execute.
phenocam_par <- model_calibration(
model = "TT",
data = phenocam_DB,
method = "GenSA",
control = list(max.call = 4000),
par_ranges = sprintf("%s/extdata/parameter_ranges.csv", path.package("phenor")),
plot = TRUE)
##
## Call:
## stats::lm(formula = data$transition_dates ~ out)
##
## Residuals:
## Min 1Q Median 3Q Max
## -24.311 -5.321 -1.247 4.821 35.776
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 0.0009523 4.9037867 0.00 1
## out 0.9933004 0.0397814 24.97 <2e-16 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 8.737 on 356 degrees of freedom
## Multiple R-squared: 0.6365, Adjusted R-squared: 0.6355
## F-statistic: 623.4 on 1 and 356 DF, p-value: < 2.2e-16
# you can specify or alter the parameter ranges as located in
# copy this file and use the par_ranges parameter to use your custom version
print(sprintf("%s/extdata/parameter_ranges.csv", path.package("phenor")))
## [1] "/Library/Frameworks/R.framework/Versions/3.6/Resources/library/phenor/extdata/parameter_ranges.csv"
We can list the parameters by looking at one of the nested list items (par).
# only list the TT model parameters, ignore other
# ancillary fields
print(phenocam_par$par)
## [1] 176.35246 -4.39729 549.56298
Phenology Model Predictions
To finally evaluate how these results would change phenology by the end of the
century we use the formatted CMIP5 data to estimate_phenology() with those
given drivers.
We will use demo CMIP5 data, instead of the data we downloaded earlier, so that
our model comes processes faster.
# download the cmip5 files from the demo repository
download.file("https://github.com/khufkens/phenocamr_phenor_demo/raw/master/data/phenor_cmip5_data_MIROC5_2090_rcp85.rds",
"phenor_cmip5_data_MIROC5_2090_rcp85.rds")
download.file("https://github.com/khufkens/phenocamr_phenor_demo/raw/master/data/phenor_cmip5_data_MIROC5_2010_rcp85.rds",
"phenor_cmip5_data_MIROC5_2010_rcp85.rds")
# read in cmip5 data
cmip5_2090 <- readRDS("phenor_cmip5_data_MIROC5_2090_rcp85.rds")
cmip5_2010 <- readRDS("phenor_cmip5_data_MIROC5_2010_rcp85.rds")
Now that we have both the phenocam data and the climate date we want run our
model projection.
# project results forward to 2090 using the phenocam parameters
cmip5_projection_2090 <- phenor::estimate_phenology(
par = phenocam_par$par, # provide parameters
data = cmip5_2090, # provide data
model = "TT" # make sure to use the same model !
)
# project results forward to 2010 using the phenocam parameters
cmip5_projection_2010 <- phenor::estimate_phenology(
par = phenocam_par$par, # provide parameters
data = cmip5_2010, # provide data
model = "TT" # make sure to use the same model !
)
If data are gridded data, the output will automatically be formatted as raster
data, which can be plotted using the raster package as a map.
Let's view our model.
# plot the gridded results and overlay
# a world map outline
par(oma = c(0,0,0,0))
raster::plot(cmip5_projection_2090, main = "DOY")
maps::map("world", add = TRUE)
Maybe more intersting is showing the difference between the start (2010) and the
end (2090) of the century.
# plot the gridded results and overlay
# a world map outline for reference
par(oma = c(0,0,0,0))
raster::plot(cmip5_projection_2010 - cmip5_projection_2090,
main = expression(Delta * "DOY"))
maps::map("world", add = TRUE)
What can you take away from these model visualizations?
PEP725 data
To get phenocam data for Europe. you will likely want to use the Pan European
Phenology Project (PEP725). This section teaching you how to access PEP725 data.
PEP725 Log In
Downloading data from the PEP725 network using phenor is more elaborate as it
requires a login
on the PEP725 website
before you can access any data.
In order to move forward with this tutorial, create a login on the PEP725
website and save your login details in a plain text file (.txt) containing your
email address and password on the first and second line, respectively. Name this
file appropriately (e.g., pep725_credentials.txt.)
PEP725 Data Availability
To download PEP725 data you need to find out which data are available. You can
either consult the data portal of the website, or use the check_pep725_species()
function. This function allows you to either list all species in the dataset, or
search by (partial) matches on the species names.
# to list all species use
species_list <- phenor::check_pep725_species(list = TRUE)
## Warning in xml2::read_html(data_selection): restarting interrupted promise evaluation
## Warning in xml2::read_html(data_selection): internal error -3 in R_decompress1
## Error in xml2::read_html(data_selection): lazy-load database '/Library/Frameworks/R.framework/Versions/3.6/Resources/library/xml2/R/xml2.rdb' is corrupt
# to search only for Quercus (oak) use
quercus_nr <- phenor::check_pep725_species(species = "quercus")
## Warning in xml2::read_html(data_selection): restarting interrupted promise evaluation
## Warning in xml2::read_html(data_selection): internal error -3 in R_decompress1
## Error in xml2::read_html(data_selection): lazy-load database '/Library/Frameworks/R.framework/Versions/3.6/Resources/library/xml2/R/xml2.rdb' is corrupt
# return results
head(species_list)
## Error in head(species_list): object 'species_list' not found
head(quercus_nr)
## Error in head(quercus_nr): object 'quercus_nr' not found
A query for Quercus returns a species ID number of 111. Once you have
established the required species number you can move forward and download the species data.
The data use policy does not allow to distribute data so this will conclude
the tutorial portion on downloading PEP725 observational data. However, the use
of the formatting functions required in phenor is consistent and the example
using PhenoCam data, above, should make you confident in processing data
from the PEP725 database once downloaded.
PEP Climate Data
For the formatting of the PEP725 data, no automated routine is provided due to
the size of the download and policy of the E-OBS dataset. Register and download the
E-OBS data
for the 0.25 degree regular grid for the best estimates of TG, TN, TX, RR,
PP (0.5 degree data is supported but not recommended).
Format PEP Climate Data
Similarly, the PEP725 data have a dedicated formatting function in the phenor
package, format_pep725(). However, it will use the previously downloaded E-OBS
data to provided the required climate data for the downloaded PEP725 data
(both file directories are requested). In addition, you need to specify which
BBCH-scale value
you would like to see included in the final formatted dataset.
# provisional query, code not run due to download / login requirements
pep725_data <- phenor::format_pep725(
pep_path = ".",
eobs_path = "/your/eobs/path/",
bbch = "11",
offset = 264,
count = 60,
resolution = 0.25
)
During the NEON Data Institute, you will share the code that you create daily
with everyone on the NEONScience/DI-NEON-participants repo.
Through this week’s tutorials, you have learned the basic skills needed to
successfully share your work at the Institute including how to:
Create your own GitHub user account,
Set up Git on your computer (please do this on the computer you will be
bringing to the Institute), and
Create a Markdown file with a biography of yourself and the project you are
interested in working on at the Institute. This biography was shared with the
group via the Data Institute’s GitHub repo.
Checklist for this week’s Assignment:
You should have completed the following after Pre-institute week 2:
Fork & clone the NEON-DataSkills/DI-NEON-participants repo.
Create a .md file in the participants/2018-RemoteSensing/pre-institute2-git directory of the
repo. Name the document LastName-FirstName.md.
Write a biography that introduces yourself to the other participants. Please
provide basic information including:
name,
domain of interest,
one goal for the course,
an updated version of your Capstone Project idea,
and the list of data (NEON or other) to support the project that you created
during last week’s materials.
Push the document from your local computer to your GithHub repo.
Created a Pull Request to merge this document back into the
NEON-DataSkills/DI-NEON-participants repo.
NOTE: The Data Institute repository is a public repository, so all members of
the Institute, as well as anyone in the general public who stumbles on the repo,
can see the information. If you prefer not to share this information publicly,
please submit the same document but use a pseudonym (cartoon character names
would work well) and email us with the pseudonym so that we can connect the
submitted document to you.
Have questions? No problem. Leave your question in the comment box below.
It's likely some of your colleagues have the same question, too! And also
likely someone else knows the answer.
We've forked (made an individual copy of) the NEONScience/DI-NEON-participants repo to
our github.com account.
We've cloned the forked repo - making a copy of it on our local computers.
We've added files and content to our local copy of the repo and committed
the changes.
We've pushed those changes back up to our forked repo on github.com.
Once you've forked and cloned a repo, you are all setup to work on your project.
You won't need to repeat those steps.
When you want to add materials from your repo to the central repo,
you will use a Pull Request. LEFT: Initial workflow after you fork and clone
a repo. RIGHT: Typical workflow once a repo is established (see Git 07 tutorial). Both use pull
requests.
Source: National Ecological Observatory Network (NEON)
In this tutorial, we will learn how to transfer changes from our forked
repo in our github.com account to the central NEON Data Institute repo. Adding
information from your forked repo to the central repo in GitHub is done using a
pull request.
LEFT: To sync changes made and committed to the repo from your
local computer, you will first push the changes from your
local repo to your fork on github.com. RIGHT: Then, you will submit a
Pull Request to update the central repository.
Source: National Ecological Observatory Network (NEON)
**Data Tip:**
A pull request to another repo is similar to a "push". However it allows
for a few things:
It allows you to contribute to another repo without needing administrative
privileges to make changes to the repo.
It allows others to review your changes and suggest corrections, additions,
edits, etc.
It allows repo administrators control over what gets added to
their project repo.
The ability to suggest changes to ANY (public) repo, without needing administrative
privileges is a powerful feature of GitHub. In our case, you do not have privileges
to actually make changes to the DI-NEON-participants repo. However you can
make as many changes
as you want in your fork, and then suggest that NEON add those changes to their
repo, using a pull request. Pretty cool!
Adding to a Repo Using Pull Requests
Pull Requests in GitHub
Step 1 - Start Pull Request
To start a pull request, click the pull request button on the main repo page.
Location of the Pull Request button on a fork of the NEON
Data Institute participants repo (Note, screenshot shows a previous version of
the repo, however, the button is in the same location). Source: National Ecological Observatory
Network (NEON)
Alternatively, you can click the Pull requests tab, then on this new page click the
"New pull request" button.
Step 2 - Choose Repos to Update
Select your fork to compare with NEON central repo. When you begin a pull
request, the head and base will auto-populate as follows:
base fork: NEONScience/DI-NEON-participants
head fork: YOUR-USER-NAME/DI-NEON-participants
The above pull request configuration tells Git to sync (or update) the NEON repo
with contents from your repo.
Head vs Base
Base: the repo that will be updated, the changes will be added to this repo.
Head: the repo from which the changes come.
One way to remember this is that the “head” is always ahead of the base, so
we must add from the head to the base.
Step 3 - Verify Changes
When you compare two repos in a pull request page, git will provide an overview
of the differences (diffs) between the files (if the file is a binary file, like
code. Non-binary files will just show up as a fully new file if it had any changes).
Look over the changes and make sure nothing looks surprising.
In this split view, shows the differences between the older (LEFT)
and newer (RIGHT) document. Deletions are highlighted in red and additions
are highlighted in green.
Pull request diffs view can be changed between unified and split (arrow).
Source: National Ecological Observatory Network (NEON)
Step 4 - Create Pull Request
Click the green Create Pull Request button to create the pull request.
Step 5 - Title Pull Request
Give your pull request a title and write a brief description of your changes.
When you’re done with your message, click Create pull request!
All pull requests titles should be concise and descriptive of
the content in the pull request. More detailed notes can be left in the comments
box.
Source: National Ecological Observatory Network (NEON)
Check out the repo name up at the top (in your repo and in screenshot above)
When creating the pull request you will be automatically transferred to the base
repo. Since the central repo was the base, github will automatically transfer
you to the central repo landing page.
Step 6 - Merge Pull Request
In this final step, it’s time to merge your changes in the
NEONScience/DI-NEON-participants repo.
NOTE 1: You are only able to merge a pull request in a repo that you have
permissions to!
NOTE 2: When collaborating, it is generally poor form to merge your own Pull Request,
better to tag (@username) a collaborator in the comments so they know you want
them to look at it. They can then review and, if acceptable, merge it.
To merge, your (or someone else's PR click the green "Merge Pull Request"
button to "accept" or merge the updated commits in the central repo into your
repo. Then click Confirm Merge.
We now synced our forked repo with the central NEON Repo. The next step in working
in a GitHub workflow is to transfer any changes in the central repository into
your local repo so you can work with them.
Data Institute Activity: Submit Pull Request for Week 2 Assignment
Submit a pull request containing the .md file that you created in this
tutorial-series series. Before you submit your PR, review the
Week 2 Assignment page.
To ensure you have all of the required elements in your .md file.
To submit your PR:
Repeat the pull request steps above, with the base and head switched. Your base
will be the NEON central repo and your HEAD will be YOUR forked repo:
base fork: NEONScience/DI-NEON-participants
head fork: YOUR-USER-NAME/DI-NEON-participants
When you get to Step 6 - Merge Pull Request (PR), are you able to merge the PR?
Finally, go to the NEON Central Repo page in github.com. Look for the Pull Requests
link at the top of the page. How many Pull Requests are there?
Click on the link - do you see your Pull Request?
You can only merge a PR if you have permissions in the base repo that you are
adding to. At this point you don’t have contributor permissions to the NEON repo.
Instead someone who is a contributor on the repository will need to review and
accept the request.
After completing the pull request to upload your bio markdown file, be sure
to continue on to Git 07: Updating Your Repo by Setting Up a Remote
to learn how to update your local fork and really begin
the cycle of working with Git & GitHub in a collaborative manner.
Workflow Summary
Add updates to Central Repo with Pull Request
On github.com
Button: Create New Pull Request
Set base: central Institute repo, set head: your Fork
Make sure changes are what you want to sync
Button: Create Pull Request
Add Pull Request title & comments
Button: Create Pull Request
Button: Merge Pull Request - if working collaboratively, poor style to merge
your own PR, and you only can if you have contributor permissions
Have questions? No problem. Leave your question in the comment box below.
It's likely some of your colleagues have the same question, too! And also
likely someone else knows the answer.
This tutorial reviews how to add and commit changes to a Git repo.
## Learning Objectives
At the end of this activity, you will be able to:
Add new files or changes to existing files to your repo.
Document changes using the commit command with a message describing what has changed.
Describe the difference between git add and git commit.
Sync changes to your local repository with the repostored on GitHub.com.
Use and interpret the output from the following commands:
git status
git add
git commit
git push
Additional Resources
Diagram of Git Commands
-- this diagram includes more commands than we will
learn in this series but includes all that we use for our standard workflow.
Information on branches in Git
-- we do not focus on the use of branches in Git or GitHub, however, if you want
more information on this structure, this Git documentation may be of use.
In the previous lesson, we created a markdown (.md) file in our forked version
of the DI-NEON-participants central repo. In order for Git to recognize this
new file and track it, we need to:
Add the file to the repository using git add.
Commit the file to the repository as a set of changes to the repo (in this case, a new
document with some text content) using git commit.
Push or sync the changes we've made locally with our forked repo hosted on github.com
using git push.
After a Git repo has been cloned locally, you can now work on
any file in the repo. You use git pull to pull changes in your
fork on github.com down to your computer to ensure both repos are in sync.
Edits to a file on your computer are not recognized by Git until you
"add" and "commit" them as tracked changes in your repo.
Source: National Ecological Observatory Network (NEON)
Check Repository Status -- git status
Let's first run through some basic commands to get going with Git at the command
line. First, it's always a good idea to check the status of your repository.
This allows us to see any changes that have occurred.
Do the following:
Open bash if it's not already open.
Navigate to the DI-NEON-participants repository in bash.
Type: git status.
The commands that you type into bash should look like the code below:
# Change directory
# The directory containing the git repo that you wish to work in.
$ cd ~/Documents/GitHub/neon-data-repository-2016
# check the status of the repo
$ git status
Output:
On branch master
Your branch is up-to-date with 'origin/master'.
Changes not staged for commit:
(use "git add <file>..." to update what will be committed)
(use "git checkout -- <file>..." to discard changes in working directory)
Untracked files:
(use "git add <file>..." to include in what will be committed)
_posts/ExampleFile.md
Let's make sense of the output of the git status command.
On branch master: This tells us that we are on the master branch of the
repo. Don't worry too much about branches just yet. We will work on the master branch
throughout the Data Institute.
Changes not staged for commit: This lists any file(s) that is/are currently
being tracked by Git but have new changes that need to be added for Git to track.
Untracked file: These are all new files that have never been added to or
tracked by Git.
Use git status anytime to view any untracked changes that have occurred, what
is being tracked and what is not currently being tracked.
Add a File - git add
Next, let's add the Markdown file containing our bio and short project summary
using the command git add FileName.md. Replace FileName.md with the name
of your markdown file.
# add a file, so that changes are tracked
$ git add ExampleBioFile.md
# check status again
$ git status
On branch master
Your branch is up-to-date with 'origin/master'.
Changes to be committed:
(use "git reset HEAD <file>..." to unstage)
new file: _posts/ExampleBioFile.md
Understand the output:
Changes to be committed: This lists the new files or files with changes that
have been added to the Git tracking system but need to be committed as actual changes
in the git repository history.
**Data Tip:** If you want to delete a file from your
repo, you can do so using `git rm file-name-here.fileExtension`. If you delete
a file in the finder (Mac) or Windows Explorer, you will still have to use
`git add` at the command line to tell git that a file has been removed from the
repo, and to track that "change".
Commit Changes - git commit
When we add a file in the command line, we are telling Git to recognize that
a change has occurred. The file moves to a "staging" area where Git
recognizes a change has happened but the change has not yet been formally
documented. When we want to permanently document those changes, we
commit the change. A single commit will work for all files that are currently
added to and in the Git staging area (anything in green when we check the status).
Commit Messages
When we commit a change to the Git version control system, we need to add a commit
message. This message describes the changes made in the commit. This commit
message is helpful to us when we review commit history to see what has changed
over time and when those changes occurred. Be sure that your message
covers the change.
**Data Tip:** It is good practice to keep commit messages to fewer than 50 characters.
# commit changes with message
$ git commit -m “new example file for demonstration”
[master e3cd622] new example file for demonstration
1 file changed, 56 insertions(+), 4 deletions(-)
create mode 100644 _posts/ExampleFile.md
Understand the output:
Each commit will look slightly different but the important parts include:
master xxxxxxx this is the unique identifier for this set of changes or
this commit. You will always be able to track this specific commit (this specific
set of changes) using this identifier.
_ file change, _ insertions(+), _ deletion (-) this tells us how many files
have changed and the number of type of changes made to the files including:
insertions, and deletions.
**Data Tip:**
It is a good idea to use `git status` frequently as you are working with Git
in the shell. This allows you to keep track of change that you've made and what
Git is actually tracking.
Why Add, then Commit?
You can think of Git as taking snapshots of changes over the
life of a project. git add specifies what will go in a snapshot (putting things
in the staging area), and git commit then actually takes the snapshot and
makes a permanent record of it (as a commit). Image and caption source:
Software Carpentry
To understand what is going on with git add and git commit it is important
to understand that Git has a staging area that we add items to with git add.
Changes are not actually documented and permanently tracked until we commit them. This allows
us to commit specific groups of files at the same time if we wish. For instance,
we may decide to add and commit all R scripts together. And Markdown files in another,
separate commit.
Transfer Changes (Commits) from a Local Repo to a GitHub Repo - git push
When we are done editing our files and have committed the changes locally, we
are ready to transfer or sync these changes to our forked repo on github.com. To
do this we need to push our changes from the local Git version control to the
remote GitHub repo.
To sync local changes with github.com, we can do the following:
Check the status of our repo using git status. Are all of the changes added
and committed to the repo?
Use git push origin master. origin tells Git to push the files to the
originating repo which in this case - is our fork on github.com which we originally
cloned to our local computer. master is the repo branch that you are
currently working on.
**Data Tip:**
Note about branches in Git: We won't cover branches in these tutorials, however,
a Git repo can consist of many branches. You can think about a branch, like
an additional copy of a repo where you can work on changes and updates.
Let's push the changes that we made to the local version of our Git repo to our
fork, in our github.com account.
# check the repo status
$ git status
On branch master
Your branch is ahead of 'origin/master' by 1 commit.
(use "git push" to publish your local commits)
# transfer committed changes to the forked repo
git push origin master
Counting objects: 1, done.
Delta compression using up to 4 threads.
Compressing objects: 100% (6/6), done.
Writing objects: 100% (6/6), 1.51 KiB | 0 bytes/s, done.
Total 6 (delta 4), reused 0 (delta 0)
To https://github.com/mjones01/DI-NEON-participants.git
5022aca..e3cd622 master -> master
NOTE: You may be asked for your username and password! This is your github.com
username and password.
Understand the output:
Pay attention to the repository URL - the "origin" is the
repository that the commit was pushed to, here https://github.com/mjones01/DI-NEON-participants.git.
Note that because this repo is a fork, your URL will have your GitHub username
in it instead of "mjones01".
**Data Tip:** You can use Git and connect to GitHub
directly in the RStudio interface. If interested, read
this R-bloggers How-To.
View Commits in GitHub
Let’s view our recent commit in our forked repo on GitHub.
Go to github.com and navigate to your forked Data Institute repo - DI-NEON-participants.
Click on the commits link at the top of the page.
Look at the commits - do you see your recent commit message that you typed
into bash on your computer?
Next, click on the <>CODE link which is ABOVE the commits link in github.
Is the Markdown file that you added and committed locally at the command
line on your computer, there in the same directory (participants/pre-institute2-git) that you saved it on your
laptop?
An example .md file located within the
participants/2017-RemoteSensing/pre-institute2-git of a Data Institute repo fork.
Source: National Ecological Observatory Network (NEON)
Is Your File in the NEON Central Repo Yet?
Next, do the following:
Navigate to the NEON central
NEONScience/DI-NEON-participants
repo. (The easiest method to do this is to click the link at the top of the page under your repo name).
Look for your file in the same directory. Is your new file there? If not, why?
Remember the structure of our workflow.
We’ve added changes from our local
repo on our computer and pushed them to our fork on github.com. But this fork
is in our individual user account, not NEONS. This fork is
separate from the central repo. Changes to a fork in our github.com account
do not automatically transfer to the central repo. We need to sync them! We will
learn how to sync these two
repos in the next tutorial
Git 06: Syncing GitHub Repos with Pull Requests .
Summary Workflow - Committing Changes
On your computer, within your local copy of the Git repo:
Create a new markdown file and edit it in your favorite text editor.
On your computer, in shell (at the command line):
git status
git add FileName
git status - make sure everything is added and ready for commit
`git commit -m “messageHere”
git push origin master
On the github.com website:
Check to make sure commit is added.
Check to see if the file that you added is visible online in your Git repo.
Have questions? No problem. Leave your question in the comment box below.
It's likely some of your colleagues have the same question, too! And also
likely someone else knows the answer.
This tutorial covers how create and format Markdown files.
## Learning Objectives
At the end of this activity, you will be able to:
Create a Markdown (.md) file using a text editor.
Use basic markdown syntax to format a document including: headers, bold and italics.
What is the .md Format?
Markdown is a human readable syntax for formatting text documents. Markdown can
be used to produce nicely formatted documents including
pdf's, web pages and more. In fact, this web page that you are reading right now
is generated from a markdown document!
In this tutorial, we will create a markdown file that documents both who you are
and also the project that you might want to work on at the NEON Data Institute.
Markdown is simple plain text, that is styled using symbols, including:
#: a header element
**: bold text
*: italic text
`: code blocks
Let's review some basic markdown syntax.
Plain Text
Plain text will appear as text in a Markdown document. You can format that
text in different ways.
For example, if we want to highlight a function or some code within a plain text
paragraph, we can use one backtick on each side of the text ( ), like this:
Here is some code. This is the backtick, or grave; not an apostrophe (on most
US keyboards it is on the same key as the tilde).
To add emphasis to other text you can use bold or italics.
Have a look at the markdown below:
The use of the highlight ( `text` ) will be reserved for denoting code.
To add emphasis to other text use **bold** or *italics*.
Notice that this sentence uses a code highlight "``", bold and italics.
As a rendered markdown chunk, it looks like this:
The use of the highlight ( text ) will be reserve for denoting code when
used in text. To add emphasis to other text use bold or italics.
Horizontal Lines (rules)
Create a rule:
***
Below is the rule rendered:
Section Headings
You can create a heading using the pound (#) sign. For the headers to render
properly there must be a space between the # and the header text.
Heading one is 1 pound sign, heading two is 2 pound signs, etc as follows:
**Data Tip:**
There are many free Markdown editors out there! The
atom.io
editor is a powerful text editor package by GitHub, that also has a Markdown
renderer allowing you to see what your Markdown looks like as you are working.
## Activity: Create A Markdown Document
Now that you are familiar with the Markdown syntax, use it to create
a brief biography that:
Introduces yourself to the other participants.
Documents the project that you have in mind for the Data Institute.
Add Your Bio
First, create a .md file using the text editor of your preference. Name the
file with the naming convention:
LastName-FirstName.md
Save the file to the participants/2017-RemoteSensing/pre-institute2-git directory in your
local DI-NEON-participants repo (the copy on your computer).
Add a brief bio using headers, bold and italic formatting as makes sense.
In the bio, please provide basic information including:
Your Name
Domain of interest
One goal for the course
Add a Capstone Project Description
Next, add a revised Capstone Project idea to the Markdown document using the
heading ## Capstone Project. Be sure to specify in the document the types of
data that you think you may require to complete your project.
NOTE: The Data Institute repository is a public repository visible to anyone
with internet access. If you prefer to not share your bio information publicly,
please submit your Markdown document using a pseudonym for your name. You may also
want to use a pseudonym for your GitHub account. HINT: cartoon character names work well.
Please email us with the pseudonym so that we can connect the submitted document to you.
Got questions? No problem. Leave your question in the comment box below.
It's likely some of your colleagues have the same question, too! And also
likely someone else knows the answer.
This tutorial covers how to clone a github.com repo to your computer so
that you can work locally on files within the repo.
## Learning Objectives
At the end of this activity, you will be able to:
Be able to use the git clone command to create a local version of a GitHub
repository on your computer.
Additional Resources
Diagram of Git Commands
-- this diagram includes more commands than we will cover in this series but
includes all that we use for our standard workflow.
In the previous tutorial, we used the github.com interface to fork the central NEON repo.
By forking the NEON repo, we created a copy of it in our github.com account.
When you fork a repository on the github.com website, you are creating a
duplicate copy of it in your github.com account. This is useful as a backup
of the material. It also allows you to edit the material without modifying
the original repository.
Source: National Ecological Observatory Network (NEON)
Now we will learn how to create a local version of our forked repo on our
laptop, so that we can efficiently add to and edit repo content.
When you clone a repository to your local computer, you are creating a
copy of that same repo locally on your computer. This
allows you to edit files on your computer. And, of course, it is also yet another
backup of the material!
Source: National Ecological Observatory Network (NEON)
Copy Repo URL
Start from the github.com interface:
Navigate to the repo that you want to clone (copy) to your computer --
this should be YOUR-USER-NAME/DI-NEON-participants.
Click on the Clone or Download dropdown button and copy the URL of the repo.
The clone or download drop down allows you to copy the URL that
you will need to clone a repository. Download allows you to download a .zip file
containing all of the files in the repo.
Source: National Ecological Observatory Network (NEON).
Then on your local computer:
Your computer should already be setup with Git and a bash shell interface.
If not, please refer to the Institute setup materials before continuing.
Open bash on your computer and navigate to the local GitHub directory that
you created using the Set-up Materials.
To do this, at the command prompt, type:
$ cd ~/Documents/GitHub
Note: If you have stored your GitHub directory in a location that is different
i.e. it is not /Documents/GitHub, be sure to adjust the above code to
represent the actual path to the GitHub directory on your computer.
Now use git clone to clone, or create a copy of, the entire repo in the
GitHub directory on your computer.
# clone the forked repo to our computer
$ git clone https://github.com/neon/DI-NEON-participants.git
**Data Tip:**
Are you a Windows user and are having a hard time copying the URL into shell?
You can copy and paste in the shell environment **after** you
have the feature turned on. Right click on your bash shell window (at the top)
and select "properties". Make sure "quick edit" is checked. You should now be
able to copy and paste within the bash environment.
The output shows you what is being cloned to your computer.
Note: The output numbers that you see on your computer, representing the total file
size, etc, may differ from the example provided above.
View the New Repo
Next, let's make sure the repository is created on your
computer in the location where you think it is.
At the command line, type ls to list the contents of the current
directory.
# view directory contents
$ ls
Next, navigate to your copy of the data institute repo using cd or change
directory:
# navigate to the NEON participants repository
$ cd DI-NEON-participants
# view repository contents
$ ls
404.md _includes code
ISSUE_TEMPLATE.md _layouts images
README.md _posts index.md
_config.yml _site institute-materials
_data assets org
Alternatively, we can view the local repo DI-NEON-participants in a finder (Mac)
or Windows Explorer (Windows) window. Simply open your Documents in a window and
navigate to the new local repo.
Using either method, we can see that the file structure of our cloned repo
exactly mirrors the file structure of our forked GitHub repo.
**Thought Question:**
Is the cloned version of this repo that you just created on your laptop, a
direct copy of the NEON central repo -OR- of your forked version of the NEON
central repo?
Summary Workflow -- Create a Local Repo
In the github.com interface:
Copy URL of the repo you want to work on locally
In shell:
git clone URLhere
Note: that you can copy the URL of your repository directly from GitHub.
Got questions? No problem. Leave your question in the comment box below.
It's likely some of your colleagues have the same question, too! And also
likely someone else knows the answer.
In this tutorial, we will fork, or create a copy in your github.com account,
an existing GitHub repository. We will also explore the github.com interface.
## Learning Objectives
At the end of this activity, you will be able to:
Create a GitHub account.
Know how to navigate to and between GitHub repositories.
Create your own fork, or copy, a GitHub repository.
Explain the relationship between your forked repository and the master
repository it was created from.
Additional Resources
Diagram of Git Commands
-- this diagram includes more commands than we will
learn in this series but includes all that we use for our standard workflow.
If you do not already have a GitHub account, go to GitHub and sign up for
your free account. Pick a username that you like! This username is what your
colleagues will see as you work with them in GitHub and Git.
Take a minute to setup your account. If you want to make your account more
recognizable, be sure to add a profile picture to your account!
If you already have a GitHub account, simply sign in.
**Data Tip:** Are you a student? Sign up for the
Student Developer Pack
and get the Git Personal account free (with unlimited private repos and other
discounts/options; normally $7/month).
Navigate GitHub
Repositories, AKA Repos
Let's first discuss the repository or "repo". (The cool kids say repo, so we will
jump on the git cool kid bandwagon) and use "repo" from here on in. According to
the GitHub glossary:
A repository is the most basic element of GitHub. They're easiest to imagine
as a project's folder. A repository contains all of the project files (including
documentation), and stores each file's revision history. Repositories can have
multiple collaborators and can be either public or private.
Once you have found the Data Institute participants repo, take 5 minutes
to explore it.
Git Repo Names
First, get to know the repository naming convention. Repository names all take
the format:
OrganizationName/RepositoryName
So the full name of our repository is:
NEONScience/DI-NEON-participants
Header Tabs
At the top of the page you'll notice a series of tabs. Please focus
on the following 3 for now:
Code: Click here to view structure & contents of the repo.
Issues: Submit discussion topics, or problems that you are having with
the content in the repo, here.
Pull Requests: Submit changes to the repo for review /
acceptance. We will explore pull requests more in the
Git 06 tutorial.
Screenshot of the NEON Data Institute central repository (note,
there has been a slight change in the repo name).
The github.com search bar is at the top of the page. Notice there are 6
"tabs" below the repo name including: Code, Issues, Pull Request, Pulse,
Graphics and Settings. NOTE: Because you are not an administrator for this
repo, you will not see the "Settings" tab in your browser.
Source: National Ecological Observatory Network (NEON)
Other Text Links
A bit further down the page, you'll notice a few other links:
commits: a commit is a saved and documented change to the content
or structure of the repo. The commit history contains all changes that
have been made to that repo. We will discuss commits more in
Git 05: Git Add Changes -- Commits .
Fork a Repository
Next, let's discuss the concept of a fork on the github.com site. A fork is a
copy of the repo that you create in your account. You can fork any repo at
any time by clicking the fork button in the upper right hand corner on github.com.
Click on the "Fork" button to fork any repo. Source:
GitHub Guides.
When we fork a repo in github.com, we are telling Git to create an
exact copy of the repo that we're forking in our own github.com account.
Once the repo is in our own account, we can edit it as we now own that fork.
Note that a fork is just a copy of the repo on github.com.
Source: National Ecological Observatory Network (NEON)
## Activity: Fork the NEON Data Institute Participants Repo
Create your own fork of the DI-NEON-participants now.
**Data Tip:** You can change the name of a forked
repo and it will still be connected to the central repo from which it was forked.
For now, leave it the same.
Check Out Your Data Institute Fork
Now, check out your new fork. Its name should be:
YOUR-USER-NAME/DI-NEON-participants.
It can get confusing sometimes moving between a central repo:
A good way to figure out which repo you are viewing is to look at the name of the
repo. Does it contain your username? Or your colleagues'? Or NEON's?
Your Fork vs the Central Repo
Your fork is an exact copy, or completely in sync with, the NEON central repo.
You could confirm this by comparing your fork to the NEON central repository using
the pull request option. We will learn about pull requests in
Git06: Sync GitHub Repos with Pull Requests.
For now, take our word for it.
The fork will remain in sync with the NEON central repo until:
You begin to make changes to your forked copy of the repo.
The central repository is changed or updated by a collaborator.
If you make changes to your forked repo, the changes will not be added to the
NEON central repo until you sync your fork with the NEON central repo.
Summary Workflow -- Fork a GitHub Repository
On the github.com website:
Navigate to desired repo that you want to fork.
Click Fork button.
Have questions? No problem. Leave your question in the comment box below.
It's likely some of your colleagues have the same question, too! And also
likely someone else knows the answer.
A version control system maintains a record of changes to code and other content.
It also allows us to revert changes to a previous point in time.
Many of us have used the "append a date" to a file name version
of version control at some point in our lives. Source: "Piled Higher and
Deeper" by Jorge Cham www.phdcomics.com
Types of Version control
There are many forms of version control. Some not as good:
Save a document with a new date (we’ve all done it, but it isn’t efficient)
Google Docs "history" function (not bad for some documents, but limited in scope).
Some better:
Mercurial
Subversion
Git - which we’ll be learning much more about in this series.
**Thought Question:** Do you currently implement
any form of version control in your work?
More Resources:
Visit the version control Wikipedia list of version control platforms.
Version control facilitates two important aspects of many scientific workflows:
The ability to save and review or revert to previous versions.
The ability to collaborate on a single project.
This means that you don’t have to worry about a collaborator (or your future self)
overwriting something important. It also allows two people working on the same
document to efficiently combine ideas and changes.
**Thought Questions:** Think of a specific time when
you weren’t using version control that it would have been useful.
Why would version control have been helpful to your project & work flow?
What were the consequences of not having a version control system in place?
How Version Control Systems Works
Simple Version Control Model
A version control system keeps track of what has changed in one or more files
over time. The way this tracking occurs, is slightly different between various
version control tools including git, mercurial and svn. However the
principle is the same.
Version control systems begin with a base version of a document. They then
save the committed changes that you make. You can think of version control
as a tape: if you rewind the tape and start at the base document, then you can
play back each change and end up with your latest version.
A version control system saves changes to a document, sequentially,
as you add and commit them to the system.
Source: Software Carpentry
Once you think of changes as separate from the document itself, you can then
think about “playing back” different sets of changes onto the base document.
You can then retrieve, or revert to, different versions of the document.
The benefit of version control when you are in a collaborative environment is that
two users can make independent changes to the same document.
Different versions of the same document can be saved within a
version control system.
Source: Software Carpentry
If there aren’t conflicts between the users changes (a conflict is an area
where both users modified the same part of the same document in different
ways) you can review two sets of changes on the same base document.
Two sets of changes to the same base document can be reviewed
together, within a version control system if there are no conflicts (areas
where both users modified the same part of the same document in different ways).
Changes submitted by both users can then be merged together.
Source: Software Carpentry
A version control system is a tool that keeps track of these changes for us.
Each version of a file can be viewed and reverted to at any time. That way if you
add something that you end up not liking or delete something that you need, you
can simply go back to a previous version.
Git & GitHub - A Distributed Version Control Model
GitHub uses a distributed version control model. This means that there can be
many copies (or forks in GitHub world) of the repository.
One advantage of a distributed version control system is that there
are many copies of the repository. Thus, if any server or computer dies, any of
the client repositories can be copied and used to restore the data! Every clone
(or fork) is a full backup of all the data.
Source: Pro Git by Scott Chacon & Ben Straub
Have a look at the graphic below. Notice that in the example, there is a "central"
version of our repository. Joe, Sue and Eve are all working together to update
the central repository. Because they are using a distributed system, each user (Joe,
Sue and Eve) has their own copy of the repository and can contribute to the central
copy of the repository at any time.
Distributed version control models allow many users to
contribute to the same central document.
Source: Better Explained
Create A Working Copy of a Git Repo - Fork
There are many different Git and GitHub workflows. In the NEON Data Institute,
we will use a distributed workflow with a Central Repository. This allows
us all (all of the Institute participants) to work independently. We can then
contribute our changes to update the Central (NEON) Repository. Our collaborative workflow goes
like this:
You will create a copy of this repository (known as a fork) in your own GitHub account.
You will then clone (copy) the repository to your local computer. You
will do your work locally on your laptop.
When you are ready to submit your changes to the NEON repository, you will:
Sync your local copy of the repository with NEON's central
repository so you have the most up to date version, and then,
Push the changes you made to your local copy (or fork) of the repository to
NEON's main repository.
Each participant in the institute will be contributing to the NEON central
repository using the same workflow! Pretty cool stuff.
The NEON central repository is the final working version of our
project. You can fork or create a copy of this repository
into your github.com account. You can then copy or clone your
fork, to your local computer where you can make edits. When you are done
working, you can push or transfer those edits back to your local fork. When
you are read to update the NEON central repository, you submit a pull
request. We will walk through the steps of this workflow over the
next few lessons.
Source: National Ecological Observatory Network (NEON)
Let's get some terms straight before we go any further.
Central repository - the central repository is what all participants will
add to. It is the "final working version" of the project.
Your forked repository - is a "personal” working copy of the
central repository stored in your GitHub account. This is called a fork.
When you are happy with your work, you update your repo from the central repo,
then you can update your changes to the central NEON repository.
Your local repository - this is a local version of your fork on your
own computer. You will most often do all of your work locally on your computer.
**Data Tip:** Other Workflows -- There are many other
git workflows.
Read more about other workflows.
This resource mentions Bitbucket, another web-based hosting service like GitHub.
Additional Resources:
Further documentation for and how-to-use direction for Git, is provided by the
Git Pro version 2 book by Scott Chacon and Ben Straub ,
available in print or online. If you enjoy learning from videos, the site hosts
several.
This tutorial builds upon
the previous tutorial,
to work with shapefile attributes in R and explores how to plot multiple
shapefiles using base R graphics. It then covers
how to create a custom legend with colors and symbols that match your plot.
Learning Objectives
After completing this tutorial, you will be able to:
Plot multiple shapefiles using base R graphics.
Apply custom symbology to spatial objects in a plot in R.
Customize a baseplot legend in R.
Things You’ll Need To Complete This Tutorial
You will need the most current version of R and preferably RStudio loaded
on your computer to complete this tutorial.
R Script & Challenge Code: NEON data lessons often contain challenges that reinforce
learned skills. If available, the code for challenge solutions is found in the
downloadable R script of the entire lesson, available in the footer of each lesson page.
Load the Data
To work with vector data in R, we can use the rgdal library. The raster
package also allows us to explore metadata using similar commands for both
raster and vector files.
We will import three shapefiles. The first is our AOI or area of
interest boundary polygon that we worked with in
Open and Plot Shapefiles in R.
The second is a shapefile containing the location of roads and trails within the
field site. The third is a file containing the Harvard Forest Fisher tower
location. These latter two we worked with in the
Explore Shapefile Attributes & Plot Shapefile Objects by Attribute Value in R tutorial.
# load packages
# rgdal: for vector work; sp package should always load with rgdal.
library(rgdal)
# raster: for metadata/attributes- vectors or rasters
library(raster)
# set working directory to data folder
# setwd("pathToDirHere")
# Import a polygon shapefile
aoiBoundary_HARV <- readOGR("NEON-DS-Site-Layout-Files/HARV",
"HarClip_UTMZ18", stringsAsFactors = T)
## OGR data source with driver: ESRI Shapefile
## Source: "/Users/olearyd/Git/data/NEON-DS-Site-Layout-Files/HARV", layer: "HarClip_UTMZ18"
## with 1 features
## It has 1 fields
## Integer64 fields read as strings: id
# Import a line shapefile
lines_HARV <- readOGR( "NEON-DS-Site-Layout-Files/HARV", "HARV_roads", stringsAsFactors = T)
## OGR data source with driver: ESRI Shapefile
## Source: "/Users/olearyd/Git/data/NEON-DS-Site-Layout-Files/HARV", layer: "HARV_roads"
## with 13 features
## It has 15 fields
# Import a point shapefile
point_HARV <- readOGR("NEON-DS-Site-Layout-Files/HARV",
"HARVtower_UTM18N", stringsAsFactors = T)
## OGR data source with driver: ESRI Shapefile
## Source: "/Users/olearyd/Git/data/NEON-DS-Site-Layout-Files/HARV", layer: "HARVtower_UTM18N"
## with 1 features
## It has 14 fields
Plot Data
In the
Explore Shapefile Attributes & Plot Shapefile Objects by Attribute Value in R tutorial
we created a plot where we customized the width of each line in a spatial object
according to a factor level or category. To do this, we create a vector of
colors containing a color value for EACH feature in our spatial object grouped
by factor level or category.
# view the factor levels
levels(lines_HARV$TYPE)
## [1] "boardwalk" "footpath" "stone wall" "woods road"
# create vector of line width values
lineWidth <- c(2,4,3,8)[lines_HARV$TYPE]
# view vector
lineWidth
## [1] 8 4 4 3 3 3 3 3 3 2 8 8 8
# create a color palette of 4 colors - one for each factor level
roadPalette <- c("blue","green","grey","purple")
roadPalette
## [1] "blue" "green" "grey" "purple"
# create a vector of colors - one for each feature in our vector object
# according to its attribute value
roadColors <- c("blue","green","grey","purple")[lines_HARV$TYPE]
roadColors
## [1] "purple" "green" "green" "grey" "grey" "grey" "grey"
## [8] "grey" "grey" "blue" "purple" "purple" "purple"
# create vector of line width values
lineWidth <- c(2,4,3,8)[lines_HARV$TYPE]
# view vector
lineWidth
## [1] 8 4 4 3 3 3 3 3 3 2 8 8 8
# in this case, boardwalk (the first level) is the widest.
plot(lines_HARV,
col=roadColors,
main="NEON Harvard Forest Field Site\n Roads & Trails \nLine Width Varies by Type Attribute Value",
lwd=lineWidth)
**Data Tip:** Given we have a factor with 4 levels,
we can create a vector of numbers, each of which specifies the thickness of each
feature in our `SpatialLinesDataFrame` by factor level (category): `c(6,4,1,2)[lines_HARV$TYPE]`
Add Plot Legend
In the
the previous tutorial,
we also learned how to add a basic legend to our plot.
bottomright: We specify the location of our legend by using a default
keyword. We could also use top, topright, etc.
levels(objectName$attributeName): Label the legend elements using the
categories of levels in an attribute (e.g., levels(lines_HARV$TYPE) means use
the levels boardwalk, footpath, etc).
fill=: apply unique colors to the boxes in our legend. palette() is
the default set of colors that R applies to all plots.
Let's add a legend to our plot.
plot(lines_HARV,
col=roadColors,
main="NEON Harvard Forest Field Site\n Roads & Trails\n Default Legend")
# we can use the color object that we created above to color the legend objects
roadPalette
## [1] "blue" "green" "grey" "purple"
# add a legend to our map
legend("bottomright",
legend=levels(lines_HARV$TYPE),
fill=roadPalette,
bty="n", # turn off the legend border
cex=.8) # decrease the font / legend size
However, what if we want to create a more complex plot with many shapefiles
and unique symbols that need to be represented clearly in a legend?
Plot Multiple Vector Layers
Now, let's create a plot that combines our tower location (point_HARV),
site boundary (aoiBoundary_HARV) and roads (lines_HARV) spatial objects. We
will need to build a custom legend as well.
To begin, create a plot with the site boundary as the first layer. Then layer
the tower location and road data on top using add=TRUE.
# Plot multiple shapefiles
plot(aoiBoundary_HARV,
col = "grey93",
border="grey",
main="NEON Harvard Forest Field Site")
plot(lines_HARV,
col=roadColors,
add = TRUE)
plot(point_HARV,
add = TRUE,
pch = 19,
col = "purple")
# assign plot to an object for easy modification!
plot_HARV<- recordPlot()
Customize Your Legend
Next, let's build a custom legend using the symbology (the colors and symbols)
that we used to create the plot above. To do this, we will need to build three
things:
A list of all "labels" (the text used to describe each element in the legend
to use in the legend.
A list of colors used to color each feature in our plot.
A list of symbols to use in the plot. NOTE: we have a combination of points,
lines and polygons in our plot. So we will need to customize our symbols!
Let's create objects for the labels, colors and symbols so we can easily reuse
them. We will start with the labels.
# create a list of all labels
labels <- c("Tower", "AOI", levels(lines_HARV$TYPE))
labels
## [1] "Tower" "AOI" "boardwalk" "footpath" "stone wall"
## [6] "woods road"
# render plot
plot_HARV
# add a legend to our map
legend("bottomright",
legend=labels,
bty="n", # turn off the legend border
cex=.8) # decrease the font / legend size
Now we have a legend with the labels identified. Let's add colors to each legend
element next. We can use the vectors of colors that we created earlier to do this.
# we have a list of colors that we used above - we can use it in the legend
roadPalette
## [1] "blue" "green" "grey" "purple"
# create a list of colors to use
plotColors <- c("purple", "grey", roadPalette)
plotColors
## [1] "purple" "grey" "blue" "green" "grey" "purple"
# render plot
plot_HARV
# add a legend to our map
legend("bottomright",
legend=labels,
fill=plotColors,
bty="n", # turn off the legend border
cex=.8) # decrease the font / legend size
Great, now we have a legend! However, this legend uses boxes to symbolize each
element in the plot. It might be better if the lines were symbolized as a line
and the points were symbolized as a point symbol. We can customize this using
pch= in our legend: 16 is a point symbol, 15 is a box.
**Data Tip:** To view a short list of `pch` symbols,
type `?pch` into the R console.
# create a list of pch values
# these are the symbols that will be used for each legend value
# ?pch will provide more information on values
plotSym <- c(16,15,15,15,15,15)
plotSym
## [1] 16 15 15 15 15 15
# Plot multiple shapefiles
plot_HARV
# to create a custom legend, we need to fake it
legend("bottomright",
legend=labels,
pch=plotSym,
bty="n",
col=plotColors,
cex=.8)
Now we've added a point symbol to represent our point element in the plot. However
it might be more useful to use line symbols in our legend
rather than squares to represent the line data. We can create line symbols,
using lty = (). We have a total of 6 elements in our legend:
A Tower Location
An Area of Interest (AOI)
and 4 Road types (levels)
The lty list designates, in order, which of those elements should be
designated as a line (1) and which should be designated as a symbol (NA).
Our object will thus look like lty = c(NA,NA,1,1,1,1). This tells R to only use a
line element for the 3-6 elements in our legend.
Once we do this, we still need to modify our pch element. Each line element
(3-6) should be represented by a NA value - this tells R to not use a
symbol, but to instead use a line.
# create line object
lineLegend = c(NA,NA,1,1,1,1)
lineLegend
## [1] NA NA 1 1 1 1
plotSym <- c(16,15,NA,NA,NA,NA)
plotSym
## [1] 16 15 NA NA NA NA
# plot multiple shapefiles
plot_HARV
# build a custom legend
legend("bottomright",
legend=labels,
lty = lineLegend,
pch=plotSym,
bty="n",
col=plotColors,
cex=.8)
### Challenge: Plot Polygon by Attribute
Using the NEON-DS-Site-Layout-Files/HARV/PlotLocations_HARV.shp shapefile,
create a map of study plot locations, with each point colored by the soil type
(soilTypeOr). How many different soil types are there at this particular field
site? Overlay this layer on top of the lines_HARV layer (the roads). Create a
custom legend that applies line symbols to lines and point symbols to the points.
Modify the plot above. Tell R to plot each point, using a different
symbol of pch value. HINT: to do this, create a vector object of symbols by
factor level using the syntax described above for line width:
c(15,17)[lines_HARV$soilTypeOr]. Overlay this on top of the AOI Boundary.
Create a custom legend.
In this tutorial, we will cover the R knitr package that is used to convert
R Markdown into a rendered document (HTML, PDF, etc).
Learning Objectives
At the end of this activity, you will:
Be able to produce (‘knit’) an HTML file from a R Markdown file.
Know how to modify chunk options to change the output in your HTML file.
Things You’ll Need To Complete This Tutorial
You will need the most current version of R and, preferably, RStudio loaded on
your computer to complete this tutorial.
Install R Packages
knitr:install.packages("knitr")
rmarkdown:install.packages("rmarkdown")
Share & Publish Results Directly from Your Code!
The knitr package allow us to:
Publish & share preliminary results with collaborators.
Create professional reports that document our workflow and results directly
from our code, reducing the risk of accidental copy and paste or transcription errors.
Document our workflow to facilitate reproducibility.
Efficiently change code outputs (figures, files) given changes in the data, methods, etc.
Publish from Rmd files with knitr
To complete this tutorial you need:
The R knitr package to complete this tutorial. If you need help installing
packages, visit
the R packages tutorial.
An R Markdown document that contains a YAML header, code chunks and markdown
segments. If you don't have an .Rmd file, visit
the R Markdown tutorial to create one.
**When To Knit**: Knitting is a useful exercise
throughout your scientific workflow. It allows you to see what your outputs
look like and also to test that your code runs without errors.
The time required to knit depends on the length and complexity of the script
and the size of your data.
How to Knit
Location of the knit button in RStudio in Version 0.99.486.
Source: National Ecological Observatory Network (NEON)
To knit in RStudio, click the knit pull down button. You want to use the knit HTML for this lesson.
When you click the Knit HTML button, a window will open in your console
titled R Markdown. This
pane shows the knitting progress. The output (HTML in this case) file will
automatically be saved in the current working directory. If there is an error
in the code, an error message will appear with a line number in the R Console
to help you diagnose the problem.
**Data Tip:** You can run `knitr` from the command prompt
using: `render(“input.Rmd”, “all”)`.
Activity: Knit Script
Knit the .Rmd file that you built in
the last tutorial.
What does it look like?
View the Output
R Markdown (left) and the resultant HTML (right) after knitting.
Source: National Ecological Observatory Network (NEON)
When knitting is complete, the new HTML file produced will automatically open.
Notice that information from the YAML header (title, author, date) is printed
at the top of the HTML document. Then the HTML shows the text, code, and
results of the code that you included in the RMD document.
Data Institute Participants: Complete Week 2 Assignment
Be sure to carefully check your knitr output to make sure it is rendering the
way you think it should!
When you are complete, submit your .Rmd and .html files to the
NEON Institute participants GitHub repository
(NEONScience/DI-NEON-participants).
The files will have automatically saved to your R working directory, you will
need to transfer the files to the /participants/pre-institute3-rmd/
directory and submitted via a pull request.
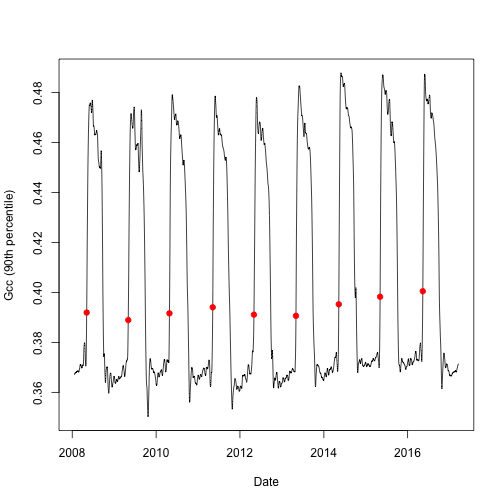
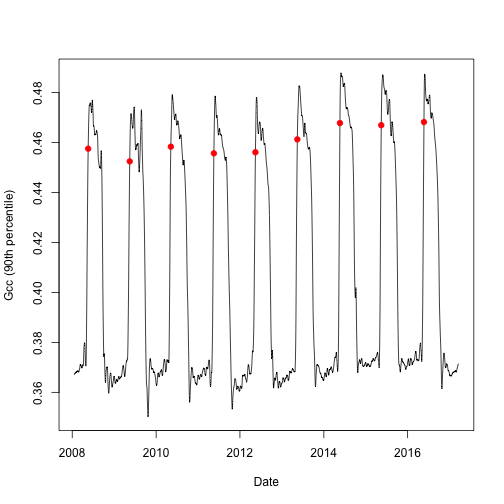
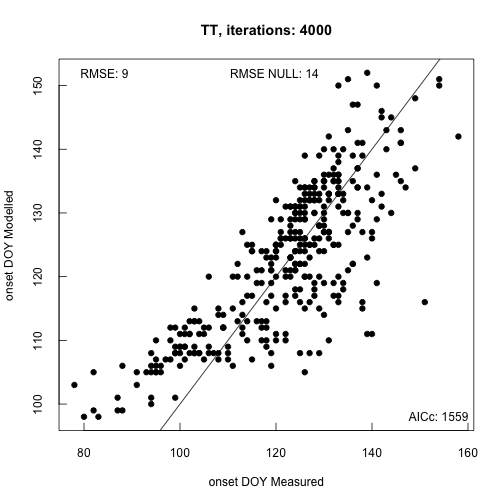
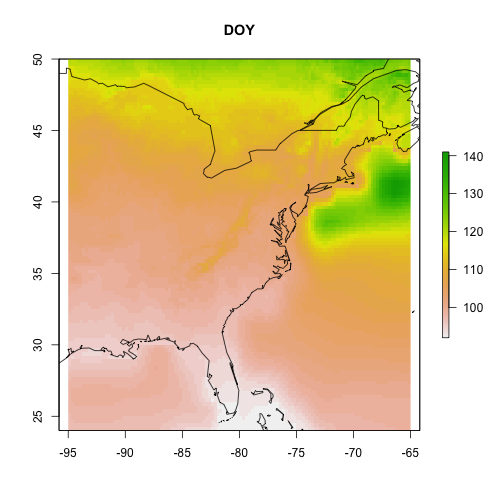
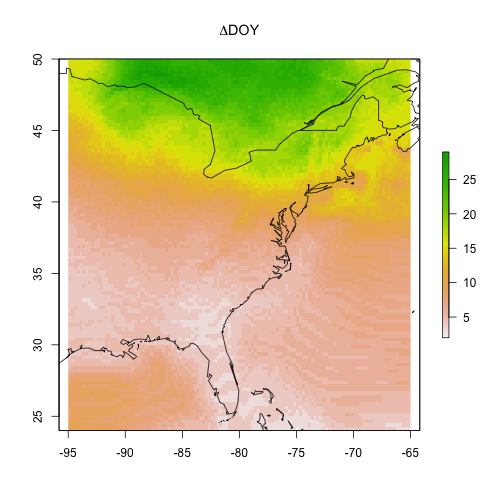
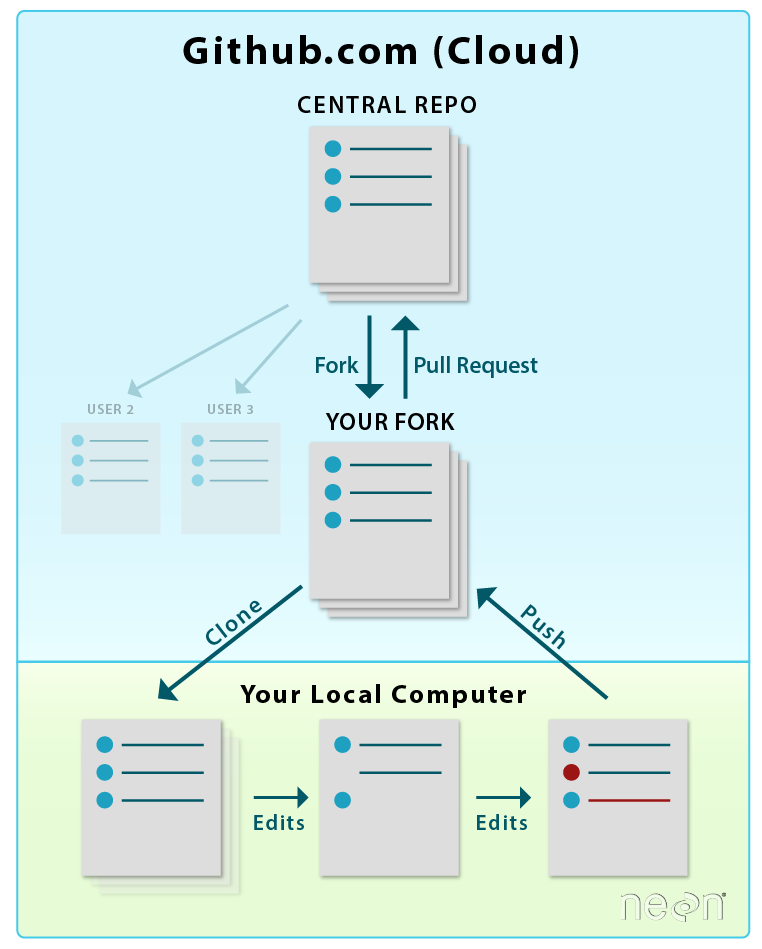

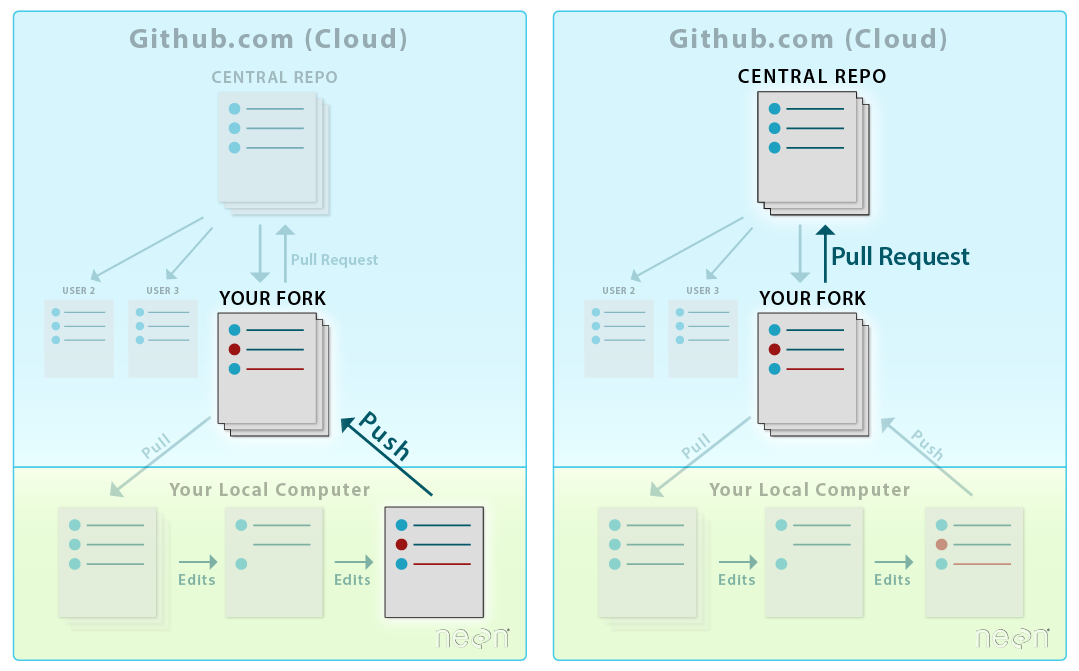
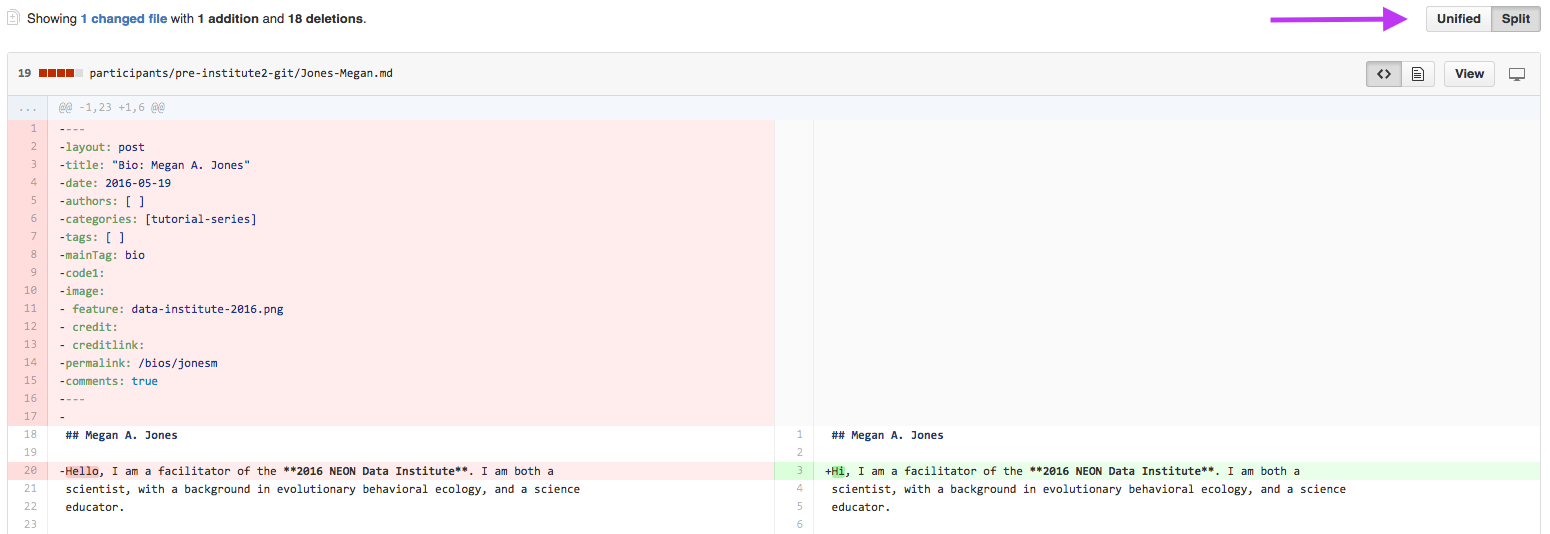
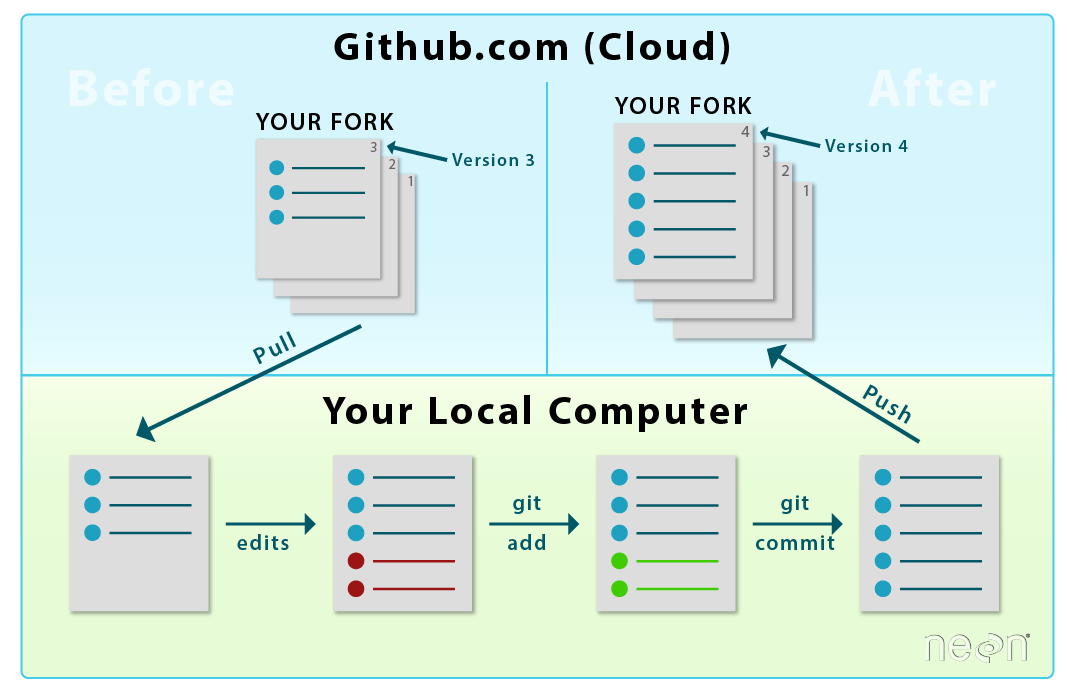

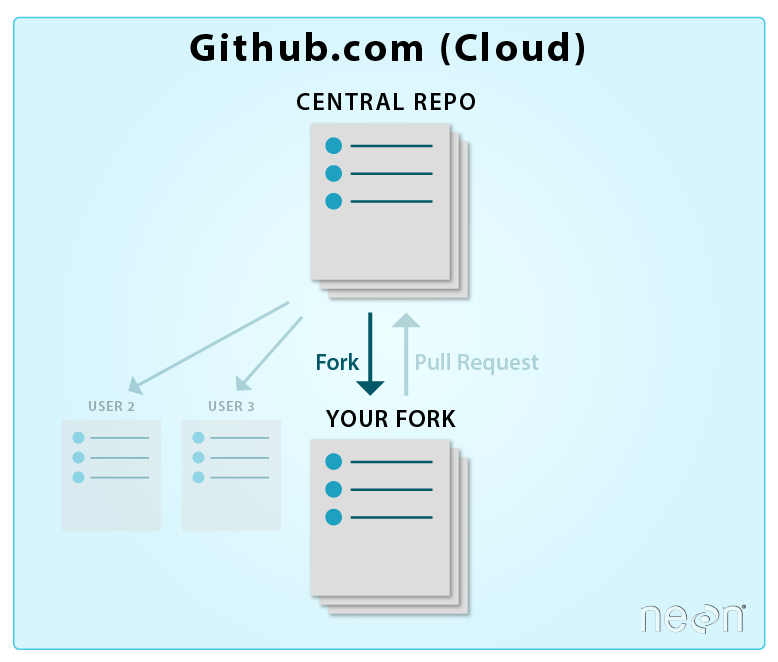
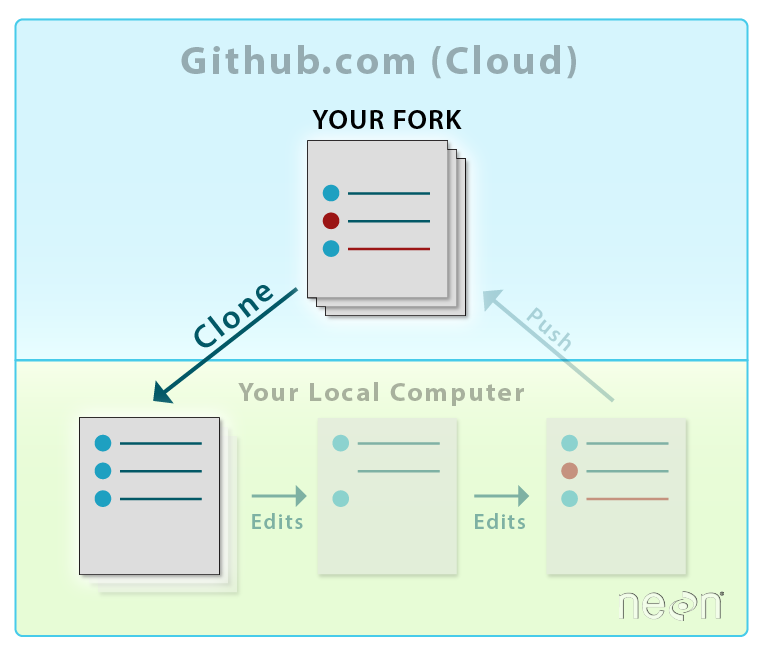




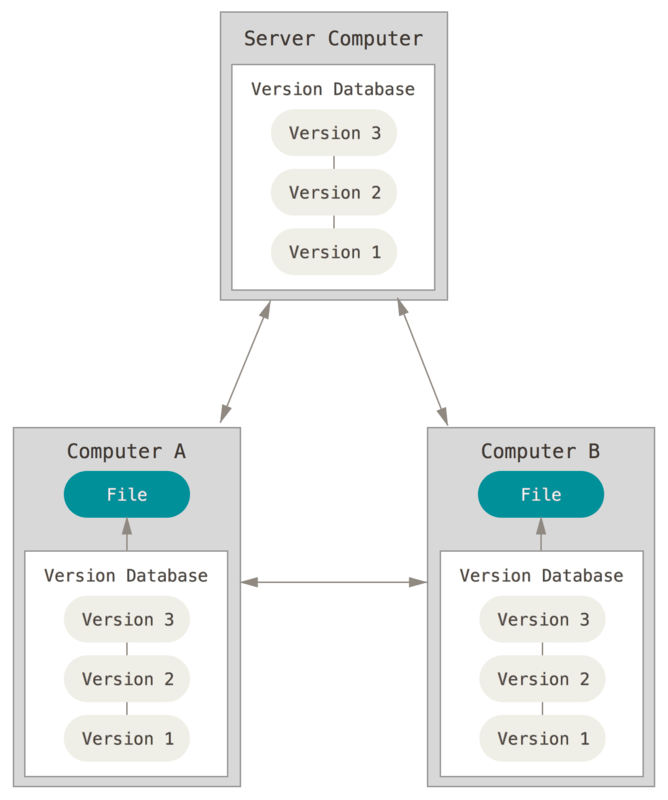
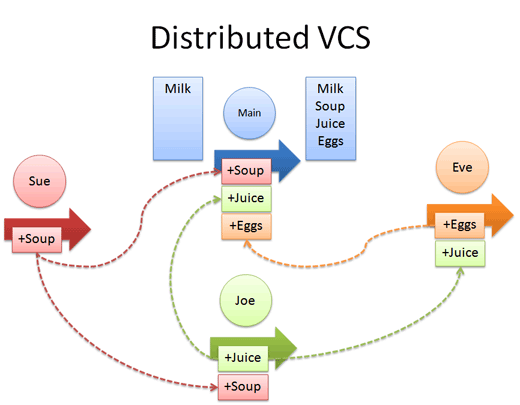
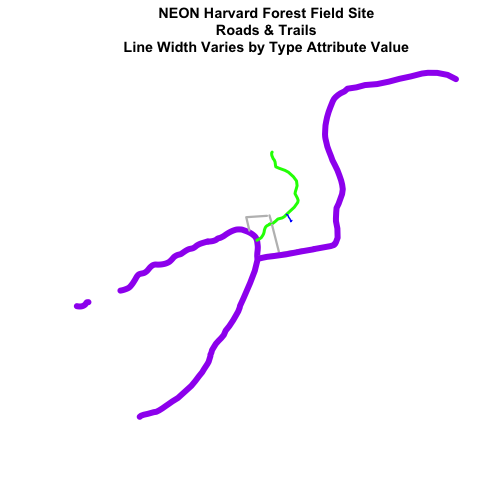
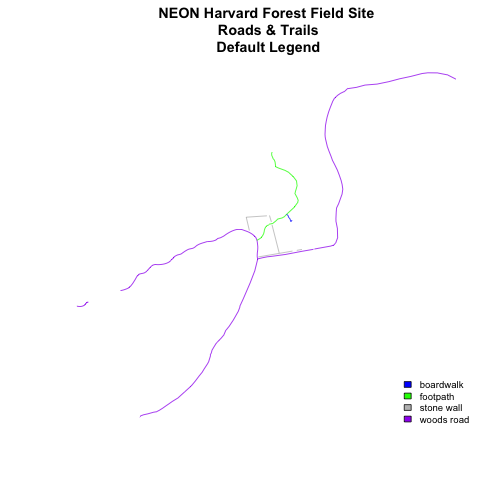




![Roads and tower location at NEON Harvard Forest Field Site with color and a modified legend varied by attribute type; each symbol on the legend corresponds to the shapefile type [i.e., tower = point, roads = lines].](https://raw.githubusercontent.com/NEONScience/NEON-Data-Skills/main/tutorials/R/Geospatial-skills/intro-vector-r/02-plot-multiple-shapefiles-custom-legend/rfigs/refine-legend-1.png)
![Roads and study plots at NEON Harvard Forest Field Site with color and a modified legend varied by attribute type; each symbol on the legend corresponds to the shapefile type [i.e., soil plots = points, roads = lines].](https://raw.githubusercontent.com/NEONScience/NEON-Data-Skills/main/tutorials/R/Geospatial-skills/intro-vector-r/02-plot-multiple-shapefiles-custom-legend/rfigs/challenge-code-plot-color-1.png)
![Roads and study plots at NEON Harvard Forest Field Site with color and a modified legend varied by attribute type; each symbol on the legend corresponds to the shapefile type [i.e., soil plots = points, roads = lines], and study plots symbols vary by soil type.](https://raw.githubusercontent.com/NEONScience/NEON-Data-Skills/main/tutorials/R/Geospatial-skills/intro-vector-r/02-plot-multiple-shapefiles-custom-legend/rfigs/challenge-code-plot-color-2.png)